
David Baker, da Universidade de Washington, e Demis Hassabis e John M. Jumper, do Google DeepMind, são os vencedores do Prêmio Nobel de Química de 2024. Metade do prêmio ficou com Baker, por sua pesquisa com design de novas proteínas, e a outra metade vai ser dividida pela dupla inglesa, pelo desenvolvimento de uma inteligência artificial capaz de prever a estrutura das proteínas.
As proteínas estão entre as moléculas mais importantes encontradas nos organismos vivos. Constituídas por cadeias de aminoácidos, estão presentes em todos os seres vivos e são responsáveis por uma infinidade de processos biológicos fundamentais para a existência de vida. “As proteínas são os motores dos nossos músculos. Elas são as máquinas que leem, copiam e reparam o DNA; são responsáveis por manter nossos neurônios sempre ativos; são os anticorpos que permitem que o nosso sistema imune responde às ameaças”, afirmou Heiner Linke, presidente do Comitê do Nobel de Química, durante o anúncio da láurea.
Na natureza existem centenas de milhares de proteínas, cada uma responsável por desempenhar diferentes funções. O papel que cada uma é capaz de desempenhar está ligado à sequência específica de aminoácidos que compõem sua estrutura. No corpo humano, por exemplo, existem 20 moléculas de aminoácidos que, combinadas de diferentes formas, geram em torno de 100 mil sequências diferentes.
A função bioquímica de cada proteína, entretanto, depende do formato ou da estrutura que essas sequências irão formar. Uma vez que a sequência se forma, as fileiras de aminoácidos se dobram no formato de estruturas tridimensionais, como o movimento de uma fita de uma ginasta rítmica congelada no tempo, ou um novelo de lã desorganizado. E são esses diferentes formatos que irão ditar o destino de cada proteína.
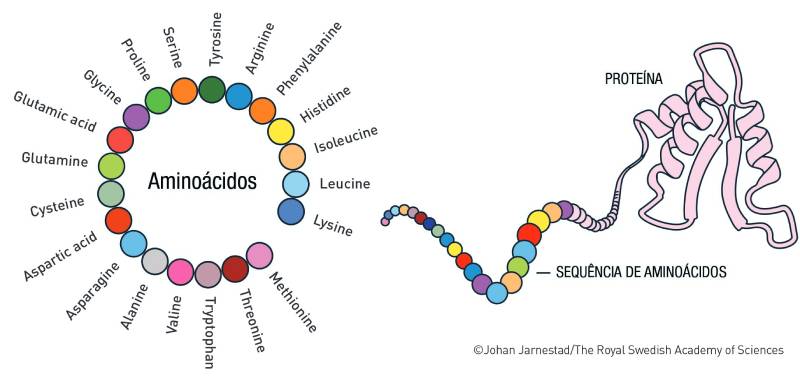
“Para entender como a vida funciona, primeiro temos que entender o formato das proteínas”, comentou Linke. Por conta disso, há muitos anos cientistas vêm buscando modos para antever o formato que será adotado por determinada proteína a partir da informação sobre as sequências de aminoácidos que a compõem. Essa capacidade se deve ao trabalho de Hassabis e Jumper, que desenvolveram uma IA capaz de prever a estrutura de praticamente qualquer proteína existente na natureza. Já Baker contribuiu para que pesquisadores do mundo inteiro fossem capazes de construir novas proteínas, com funções e formatos específicos, completamente diferentes das encontradas na natureza.
“Ao reconhecer os avanços significativos em inteligência artificial que permitiram a predição da estrutura de proteínas a partir da sequência de aminoácidos, o Nobel deste ano destaca a importância da colaboração científica interdisciplinar”, diz Douglas Moraes Mendel Soares, docente da Faculdade de Ciências Farmacêuticas da Unesp, câmpus de Araraquara. “Ao destacar os esforços conjuntos de diferentes áreas, como química, biologia e ciência da computação, o Comitê do Nobel incentiva uma abordagem mais integrada na pesquisa científica”, completa. Soares também chama a atenção para o fato desses avanços viabilizarem o desenvolvimento de proteínas específicas, que podem ser direcionadas ao tratamento de doenças e infecções, ou para a obtenção de enzimas industriais e de produtos agrícolas melhorados.
A estrutura de uma proteína
Embora o caráter fundamental das proteínas seja reconhecido desde o século 19, apenas em 1958 uma dupla de pesquisadores, John Kendrew e Max Perutz, foi capaz de tirar uma “fotografia” dessas moléculas, mostrando, pela primeira vez, que elas apresentavam uma forma tridimensional. A dupla atingiu o feito por meio de uma técnica chamada de Cristalografia de raios-X, que lhes rendeu o Prêmio Nobel de Química de 1962. Desde então, a grande possibilidade de variabilidade dessas estruturas despertou a curiosidade de pesquisadores no mundo inteiro. Foi a partir de experimentações com essas moléculas que o norte-americano Christian Anfinsen conseguiu “desenrolar” uma proteína. Porém a observação mais interessante veio na seguência: uma vez cessada a influência do pesquisador, a proteína voltou a se enrolar e assumiu exatamente o mesmo formato. Essa constatação foi um forte indício de que as moléculas não assumiam tais formatos aleatoriamente. A suposição ganhou força quando um segundo pesquisador, Cyrus Levinthal, apresentou evidências de que o formato das proteínas deveria ser pré-definido e dependia da sequência de aminoácidos que estavam em sua composição. A organização desses microcomponentes deveria conter todas as informações necessárias para entender os diferentes formatos das proteínas. Ou seja, seria possível prever a estrutura de uma proteína apenas a partir de sua sequência de aminoácidos.
Esse trabalho foi possibilitado pelo uso do método de cristalografia de Raio-X. Porém, essa ferramenta apresentava várias problemáticas que dificultavam a tarefa. “As ferramentas existentes até então se baseavam na comparação de sequências de aminoácidos com outras cuja estrutura tridimensional já havia sido caracterizada experimentalmente”, explica Soares. “Isso limitava a aplicação e, além disso, eram processos caros e demorados, pois dependiam do preparo de amostras e da aquisição de equipamentos específicos para obter os dados biológicos”, completa. Assim, apesar de, na época, ser possível caracterizar as proteínas a partir das sequências de aminoácidos, o processo se tornava ineficiente e mesmo inviável quando haviam grandes quantidades de informação – a nível de comparação, hoje são conhecidas mais de 200 milhões de proteínas.
Com o desafio de acelerar esse processo, Hassabis e Jumper decidiram aproveitar a alta capacidade de análise das Inteligências Artificiais, em particular após a criação das redes neurais, que simulavam o funcionamento do cérebro humano e permitiam que as máquinas aprendessem com novos dados e treinamento. Tendo isso com base, em 2020, a dupla lançou uma nova IA, batizada de “AlphaFold2”, capaz de alcançar 90% de acurácia ao prever qual seria o formato de uma proteína a partir de sua sequência de aminoácidos. Para alcançar tamanha exatidão, o grupo treinou a Inteligência Artificial com todas as bases de dados de proteínas existentes, feito que só foi possível graças a iniciativas de ciência aberta.
Desde 2020, o AlphaFold2 segue recebendo atualizações e melhorias e já foi utilizado por pesquisadores no mundo inteiro para prever as estruturas de milhões de proteínas que não haviam sido descritas até então. Esse avanço é particularmente importante em um cenário no qual mais e mais cientistas exploram as possibilidade de criação de novas proteínas: ao saber como as sequências de aminoácidos irão se comportar, fica mais fácil definir a função específica que se espera daquela molécula.
A combinação de diferentes peças
Mesmo antes do uso de inteligências artificiais, pesquisadores já exploravam a possibilidade de manipular os aminoácidos das proteínas como peças de lego. “A partir de alterações na sequência de aminoácidos, é possível planejar modificações na estrutura e função de proteínas, o que possibilita a criação de versões inéditas com diversas aplicações na indústria farmacêutica, na agricultura e na pesquisa científica”, explica Soares.
Estimulados por essa possibilidade, pesquisadores começaram a testar diferentes combinações de aminoácidos. No entanto, logo se depararam com uma restrição: o número de aminoácidos disponíveis na natureza é restrito, o que impôs um limite para as experimentações. Determinado a ultrapassar essa barreira, Baker propôs algo revolucionário: criar uma proteína inteiramente do zero.
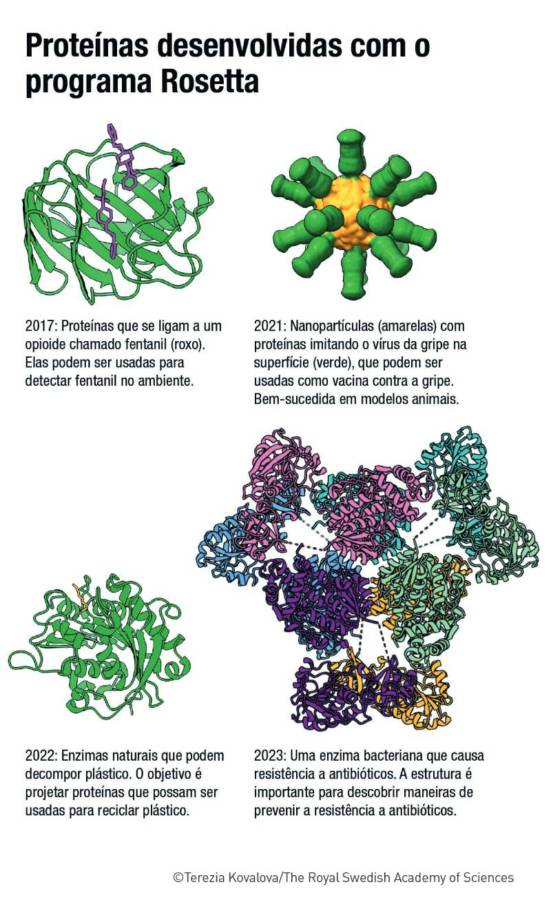
Em 1999, Baker desenvolveu um software chamado Rosetta, utilizado para estudar as estruturas das proteínas. O grupo liderado pelo norte-americano inseria sequências de aminoácidos que eram analisadas pelo programa e utilizadas para prever quais possíveis formatos a proteína teria. Apesar de ser uma ferramenta com várias limitações, ela foi útil para gerar importantes reflexões sobre os objetos de pesquisa.
O mais importante feito do Rosetta veio anos mais tarde quando, na busca por construir uma proteína do zero, Baker explorou a possibilidade de usar o software “ao contrário”. Em vez de inserir uma sequência de aminoácidos e observar o formato resultante, o grupo de pesquisadores do Departamento de Bioquímica da Universidade de Washington liderado por Baker criou uma estrutura completamente nova e fez com que o Rosetta indicasse qual seria a sequência necessária para alcançar esse resultado. Para fazer isso, o Rosetta pesquisou em um banco de dados de todas as estruturas proteicas conhecidas e procurou por pequenos fragmentos de proteínas que tivessem semelhanças com a estrutura desejada e, assim, foi construindo o manual de instruções das peças de lego necessárias.
O resultado foi testado experimentalmente pela equipe e resultou em um grande sucesso. A proteína, apelidada de Top7, apresentava praticamente a mesma estrutura prevista na etapa de planejamento. Até então, todos os que tinham se aventurado nesse campo de pesquisa lograram apenas criar proteínas imitando estruturas já existentes. O experimento de Baker marcou a primeira vez em que pesquisadores conseguiram criar uma estrutura completamente nova e que não existia na natureza, O trabalho foi publicado em 2003, quando Baker também liberou o código do Rosetta, isso permitiu o uso do programa pela comunidade científica do mundo inteiro, que vem contribuindo para o seu aperfeiçoamento e encontrando novas áreas para empregá-lo.
O futuro das proteínas
Os três laureados contribuíram de maneira definitiva para que, hoje, entendamos um pouco mais sobre a origem da vasta diversidade que existe entre os organismos vivos. Além de acelerar processos de visualização de estruturas proteicas, o que representa um impulso na ciência básica e no nosso entendimento sobre como a vida funciona, algumas doenças se desenvolvem ou mesmo como se dá a resistência a antibióticos.
Soares destaca que essas pesquisas impulsionam, também, o desenvolvimento de novas tecnologias. “No campo da medicina, pode-se desenvolver anticorpos ou vacinas que reconheçam alvos específicos e investigar enzimas que possam substituir ou complementar funções ausentes em doenças genéticas. Na agricultura, proteínas que conferem resistência a pragas podem ser identificadas e, por manipulação genética, serem inseridas em culturas de importância agronômica visando aumentos na produtividade”, explica o pesquisador.
No começo da semana foram anunciados os laureados com o Nobel de Medicina ou Fisiologia (7). A dupla de norte-americanos Victor Ambros e Gary Ruvkun recebeu a honraria pela descoberta dos microRNAs, pequenos fragmentos de RNA que controlam a atividade de genes dentro das células. Já o Nobel de Física (8) foi para John Hopfield e Geoffrey Hinton, inventores das bases que levaram à criação do aprendizado de máquina e das redes neurais artificiais. Os anúncios continuarão até a próxima segunda, com o de Literatura (10), o da Paz (11), e o de Economia (14) encerrando a série. A cerimônia de entrega dos prêmios será realizada no dia 10 de dezembro.
Ilustração de David Baker (esquerda), Demis Hassabis (meio) e John Jumper (direita). Crédito: Ill. Niklas Elmehed © Nobel Prize Outreach